
Is the SARS-CoV-2 Genome Valid?
A closer look at the first genetic reads mapping.

Abstract
Aligning the original reads that are published by Wu et al. 2020 to the SARS-CoV-2 reference genome yields a coverage of 99.993%. However, a closer look at the alignments, specifically for the head & tail of the novel genome, reveals that none of the original reads match sufficiently enough to establish certainty about the validity of the reference genome.
At the minimum it should be shown, that:
At least 10–20 reads that show a perfect alignment to the head & the tail.
A complete genetic sequence (RNA) of ~30kb length exists in the sample.
To my knowledge, neither of these two conditions have been fulfilled, to this date.
Overview of my Seven Part Genomics Series:







Background
Wu et al., 2020 was the first study publishing the original sequence. In my first post on this topic, I explained in detail how the authors worked to retrieve the first sequence. In a follow-up post, I have discussed more anomalies & open questions around the findings of the claimed viral genome of SARS-CoV-2, the alleged pathogen that causes COVID-19. The key takeaways were, that there were significant issues in reproducing the full genome via Megahit de-novo assembly, specifically, that:
The claimed longest contig, which was used as the base for the genome, to this date, has not been identically reproduced by anyone, that I am aware of.
The exact start and the complete tail of the genome was not constructed by Megahit, the assembler that produced the longest original contig.
Many studies now, do not use de-novo assembly anymore, but instead simply map the short-reads to the “reference genome”. Here’s a quick overview of the workflow:

Introduction
While the theoretical assembly via De Bruijn graph algorithm (explanatory video here) appears to be valid - for a novel genome, the edges should be specifically important. One might certainly expect, to find reads, that perfectly map to the head (and tail) of the genome - imagine it similar to an edge piece of a puzzle!

Hence, if we look at the individual reads, I would expect to find many reads, that align perfectly to the edges of the reference genome (MN908947.3).
Methods
For read alignment, I ran a handful of common bioinformatics commands, summarized in this script: ./align.sh SRR10971381 MN908947.3
Then I used the Integrative Genomics Viewer (IGV) for visualization.
Results
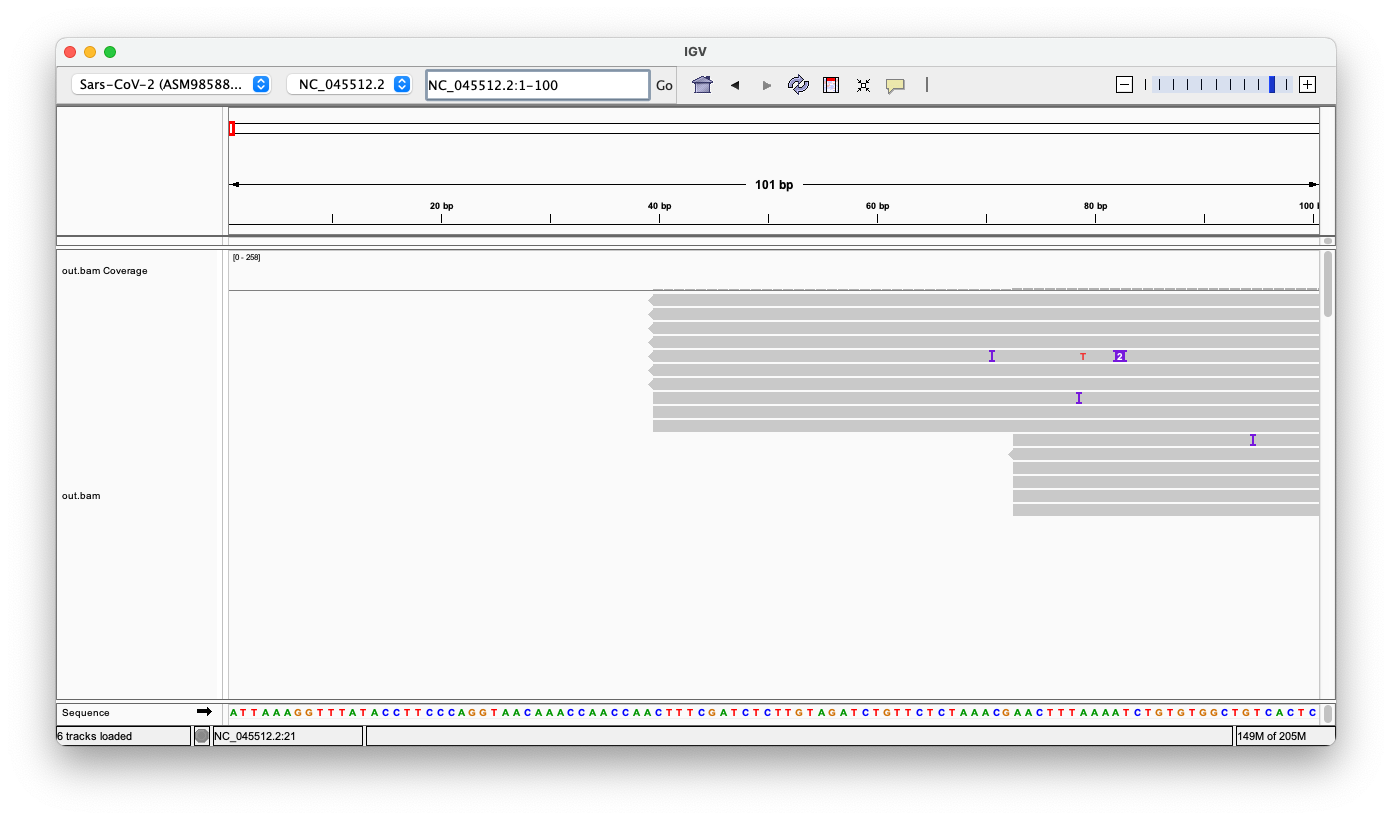
So here’s what IGV shows: The first row shows the coverage, the second all the reads, that are aligned to the reference genome (SARS-CoV-2 v3) in the third row (blue).

One can already see, that there’s a lot of highlighted coloring visible, which highlights mismatched bases, inserts and deletions, and that coverage varies vastly.
Head
Let’s look at the 'left hand/beginning' of the original Fan Wu et al. sequence.

Here are the first 64 bases. At the bottom is the official reference sequence. Only 28 reads align from the very start, seven additional reads align with less than four bases missing.
However, all reads have inserts or mismatches - not a single read fits perfectly!
For a novel genome, one might certainly expect, to find reads, that perfectly map to the start of the genome - imagine it similar to an edge piece of a puzzle!
What are mismatches?
Mismatches are when the base does not match the base at the same position in the reference genome. Inserts are inserted bases, that are added, in order to make the read align properly.

Here’s an example, based on the first read, here they’ve simply inserted ‘ATT’ at position 4, which is also highlighted by the purple icon in the above screenshot.
Tail
The tail looks even worse, as none of the read even matched the end completely. There were only two reads which came somewhat close, but still had 4-6 read errors in less than 100 bases. The short transparent/white read is actually highlighted as low-quality by IGV. I have highlighted the mismatches with a purple circle.

Discussion
None of the reads align perfectly, neither to the head nor the tail. Hence, it is unclear to me, how this sequence could have been established as the reference genome for SARS-CoV-2. To establish, that this would be indeed the reference genome, at the minimum it should be shown, that:
At least 10–20 reads that show a perfect alignment to the head & the tail.
A complete genetic sequence (RNA) of ~30kb length exists in the sample.
To my knowledge, neither of these two conditions have been fulfilled to this date.
Quality Trimmed Reads
In the above explanation, I used the reads as provided by Wu et al. 2020. It is unclear to me, if these are already quality trimmed. So I have used fastp and trimmomatic program to first quality trim, then align, but this did not lead to significantly better or convincing results. Still, it remains important to understand how the first genome could have be established with scientific certainty, that’s why focusing on the Wu et al. 2020 dataset is so important!

Full Length Sequencing Technologies
While focusing on the original Wu et al. 2020 study that establishes the reference genome is most important - a common argument for the reliability of deep metagenomic sequencing, is that there are other technologies, that can sequence longer reads (1-2kb) or possibly even the full genome.
Oxford Nanopore
Hence, I have repeated this with a Nanopore SRR as well, without any notable differences in outcome.
In almost all reads there appears to be significant issues with the alignment, leading to mismatches, inserts or deletions.
I could not find any raw reads for Nanopore long read sequencing, that also included the ends. This version, is using the Midnight protocol v3, which specifically omits the ends.



PacBio
The company PacBio claims, on their website, that they can sequence “single-molecule reads of up to 25 kb”.

They do provide sample data of their SARS-CoV-2 “HIFI sequencing” - but when looking at it in IGV, bot the tail and the head are not aligned at all (completely missing).
Conclusion
I could not find any compelling evidence, that the SARS-CoV-2 reference genome is valid, specifically when looking at the head & tail alignments.
Sources & Code
You can find all code and results mentioned in this article here:
The problem is simple - the provenance of the genetic material used to sequence the so-called sars-cov-2 genome has not been determined i.e show me the virus (isolated, purified, seperated from other biological and genetic material)
Excellent, thank you very much for this.