
The Swiss Hantavirus Sequence - What Was Actually Found?
A new Andes virus sequence from a Swiss patient linked to the MV Hondius cluster has now been posted.

A new Andes virus sequence from the Switzerland-linked outbreak has now been posted on Virological.org. That sounds like scientists directly found a full virus genome. But that is not quite what happened.
How Clinical Sequencing Works
In this kind of clinical sequencing workflow, the sample is inherently mixed. You are not starting with one clean, isolated virus genome. This is the same basic issue seen with SARS-CoV-2, where institutions such as the RKI and CDC have described sequencing workflows based on fragments from mixed clinical material rather than direct end-to-end sequencing of one complete viral genome molecule. So the starting material is a mixture of genetic material from different sources.
In one reported workflow, more than 56% of reads were confirmed human RNA after dehosting against a human genome model, and in another, about 2.9 million of 3.1 million quality-filtered reads, or roughly 93.5%, were human.

Researchers then used short-read sequencing and de Bruijn graph assembly. That means they did not read one full virus molecule from start to finish. Instead, they sequenced many short fragments, without directly knowing their full molecular origin, broke those reads into even smaller overlapping substrings called k-mers, and used those k-mers to reconstruct a longer sequence.

That can produce a plausible genome. But it still does not directly prove that this exact full sequence existed in the sample as one continuous natural molecule.
Why does that matter?
In a mixed sample, sequence pieces from different sources can be combined inside the same graph.
A de Bruijn graph shows local overlap, not one uniquely proven full biological structure. If enough short pieces fit together, the software can reconstruct a continuous sequence. But that still does not show that the exact full sequence was directly present in the sample as one intact natural molecule.
The result can therefore be a sequence that is plausible and computationally reconstructible without being direct proof that this complete viral genome was actually present as a single continuous molecule.
A Short Practical Example
I’ve built a small tool that lets you see how the de Bruijn graph method works, and why it can also go wrong.
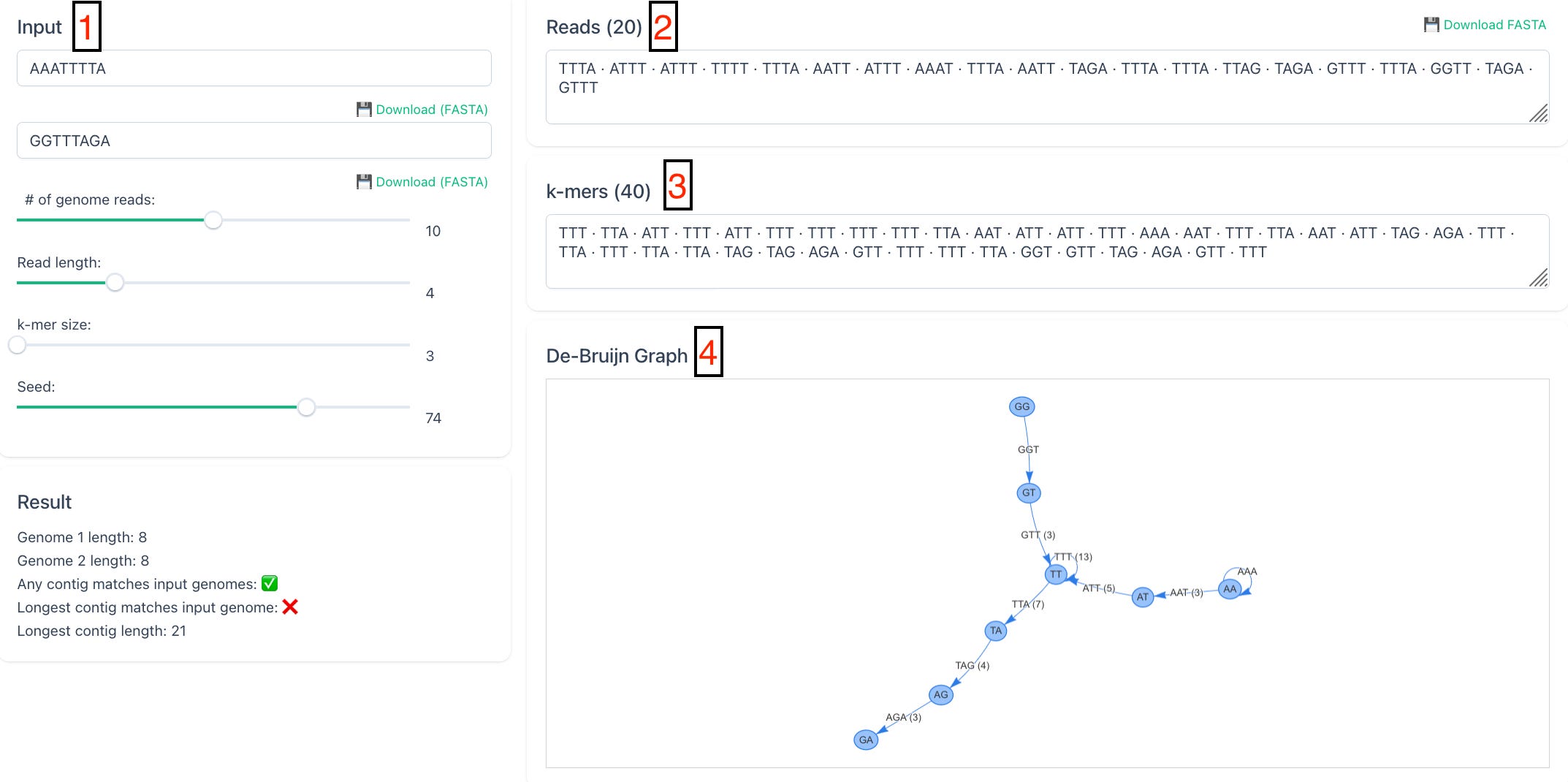
In this simplified example, we start with two assumed real input sequences.
These are then randomly chopped into short reads of length 4.
From those reads, even shorter overlapping strings called k-mers are generated, here with length 3.
A directed graph is then built from those k-mers.
From that graph, all possible contigs are assembled. The real original inputs are highlighted in green, but the graph also produces many other candidate sequences that were never part of the original input.
Later alignment to one of these candidate sequences is not proof either; it only shows compatibility with the chosen template, not direct proof that the full molecule existed in the sample.
Of course, this is a simplified example. Real-world read lengths and k-mer sizes are different, and the actual data are far more complex.
But the basic point remains the same: local overlap can generate multiple globally plausible sequences, without proving that every such contig existed as a real biological molecule.
Conclusion
So the key point is simple: a reconstructed genome is not the same thing as a directly observed complete viral molecule. It may be plausible, useful, and compatible with the reads, while still falling short of strict proof. The sequence therefore remains physically unvalidated as one directly demonstrated continuous molecule. And causality is a second-order claim: if the sequence itself is only indirectly inferred, then causality cannot be directly proven either.
Thank you very much!
Anyhow, I would not agree with the opinion that sequencing can prove that a virus or this particular virus exists.
For me, the whole hanta-story lacks logic.
If this "virus" is related to mice or rat feces: Why are these particles which some call "virus" are not phages of a typical gut bacteria of these species?
And then: Why are these bacteria not the actually pathogenic items?
Oh, I see ... because some have developed a vaccine ....
Not only would they need to rule out the possibility that a sequence was human, wouldn't they also have to rule out sequences from other organisms hosted in the human body?